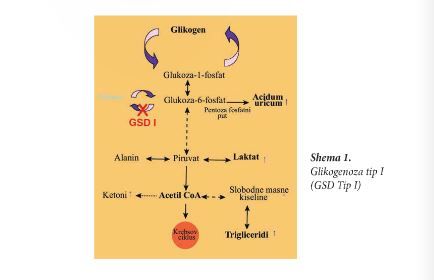
 Glikogenoza Tip 1 Tip 1 bolesti taloženja glikogena posljedica je deficita enzima glikoza-6-fosfataze koji katalizuje završni stepen u produkciji glikoze iz glikoza-6-fosfata. Deficit ovog enzima ometa produkciju glikoze iz glikogenolize i glikoneogeneze. U nedostatku glikoze nastaje postprandijalna hipoglikemija i povećana produkcija laktata, mokraćne kisjeline i triglicerida. Glikogen i neutralne masti se deponuju u jetri, što dovodi do izražene hepatomegalije. Shema 1 Glikogenoza tip I (GSD Tip I) Sistem enzima glikoza-6-fosfataze obuhvata nekoliko subjedinica. Katalitička subjedinica katalizuje završnu reakciju glikogenolize i glikoneogeneze konverziju glikoza-6-fosfata u glikozu. Glikoza-6-fosfataza je locirana u membrani endoplazmatskog retikuluma. Aktivna strana je okrenuta prema lumenu endoplazmatskog retikuluma. Tri transportna sistema prenose supstrat, G-6-P,fosfat, anorganski ortofosfat i glikozu kroz membranu endoplazmatskog retikuluma. Specifični transporter za glikoza-6-fosfat prenosi glikoza-6-fosfat u endoplazmatski retikulum, a aktuelna saznanja ukazuju da transporter za glikoza 6- fosfat takodje prenosi fosfat van endoplazmatskog retikuluma; glikoza se iz endoplazmatskog retikuluma prenosi van, pomoću specifičnog transportera GLUT2. Više od 80% pacijenata sa glikogenozom Tip 1 imaju deficit katalitičke aktivnosti glukoza-6-fosfataze što uzrokuje tip Ia glikogenoze. U oboljelih od ovog tipa glikogenoze nadjeno je više od 70 različitih mutacija gena koji enkodira glikoza-6-fosfatazu. Ove mutacije se ne nalaze u osoba sa tipom Ib glikogenoze, koja nastaje usled oštećenja transporta glikoza-6-fosfata u lumen endoplazmatskog retikuluma zbog mutacije G6PT1 gena, što prouzrokuje deficit G6P transportera. U Tipu 1 glikogenoze, smanjena aktivnost glikoza-6-fosfataze dovodi do povećane koncentracije glikoza-6-fosfata u hepatocitima i skretanja na alternativni put sa sledećim posljedicama: – laktat acidoza (kao posljedica povećane glikolize); – hiperurikemija (kao posljedica skretanja na pentoza fosfat put čime se povećano stvara mokraćna kisjelina; – hipertrigliceridemija (kao posljedica povećane sinteze acetil koenzima A, prekurzora malonil koenzima A, koji inhibira aktivnost karnitin palmitoil transferaze 1 dovodeći do smanjenja beta oksidacije masnih kiselina i povećane lipogeneze). Glikogenoza Tip Ia Glikogenoza Tip Ia nastaje usljed mutacije glukoza-6-fosfat C gena (G6PC) koji je lociran na hromozomu 17q21 i nasledjuje se autozomalno recesivno. Prosječna incidencija je l slučaj na 100.000 rođene djece. Glikogenoza Tip Ia se dešava u svim etničkim grupama, a najčešće mutacije su u Ashkenaza Jevreja, Kineza, Japanaca i Meksikanaca
Glikogenoza Tip 1 Tip 1 bolesti taloženja glikogena posljedica je deficita enzima glikoza-6-fosfataze koji katalizuje završni stepen u produkciji glikoze iz glikoza-6-fosfata. Deficit ovog enzima ometa produkciju glikoze iz glikogenolize i glikoneogeneze. U nedostatku glikoze nastaje postprandijalna hipoglikemija i povećana produkcija laktata, mokraćne kisjeline i triglicerida. Glikogen i neutralne masti se deponuju u jetri, što dovodi do izražene hepatomegalije. Shema 1 Glikogenoza tip I (GSD Tip I) Sistem enzima glikoza-6-fosfataze obuhvata nekoliko subjedinica. Katalitička subjedinica katalizuje završnu reakciju glikogenolize i glikoneogeneze konverziju glikoza-6-fosfata u glikozu. Glikoza-6-fosfataza je locirana u membrani endoplazmatskog retikuluma. Aktivna strana je okrenuta prema lumenu endoplazmatskog retikuluma. Tri transportna sistema prenose supstrat, G-6-P,fosfat, anorganski ortofosfat i glikozu kroz membranu endoplazmatskog retikuluma. Specifični transporter za glikoza-6-fosfat prenosi glikoza-6-fosfat u endoplazmatski retikulum, a aktuelna saznanja ukazuju da transporter za glikoza 6- fosfat takodje prenosi fosfat van endoplazmatskog retikuluma; glikoza se iz endoplazmatskog retikuluma prenosi van, pomoću specifičnog transportera GLUT2. Više od 80% pacijenata sa glikogenozom Tip 1 imaju deficit katalitičke aktivnosti glukoza-6-fosfataze što uzrokuje tip Ia glikogenoze. U oboljelih od ovog tipa glikogenoze nadjeno je više od 70 različitih mutacija gena koji enkodira glikoza-6-fosfatazu. Ove mutacije se ne nalaze u osoba sa tipom Ib glikogenoze, koja nastaje usled oštećenja transporta glikoza-6-fosfata u lumen endoplazmatskog retikuluma zbog mutacije G6PT1 gena, što prouzrokuje deficit G6P transportera. U Tipu 1 glikogenoze, smanjena aktivnost glikoza-6-fosfataze dovodi do povećane koncentracije glikoza-6-fosfata u hepatocitima i skretanja na alternativni put sa sledećim posljedicama: – laktat acidoza (kao posljedica povećane glikolize); – hiperurikemija (kao posljedica skretanja na pentoza fosfat put čime se povećano stvara mokraćna kisjelina; – hipertrigliceridemija (kao posljedica povećane sinteze acetil koenzima A, prekurzora malonil koenzima A, koji inhibira aktivnost karnitin palmitoil transferaze 1 dovodeći do smanjenja beta oksidacije masnih kiselina i povećane lipogeneze). Glikogenoza Tip Ia Glikogenoza Tip Ia nastaje usljed mutacije glukoza-6-fosfat C gena (G6PC) koji je lociran na hromozomu 17q21 i nasledjuje se autozomalno recesivno. Prosječna incidencija je l slučaj na 100.000 rođene djece. Glikogenoza Tip Ia se dešava u svim etničkim grupama, a najčešće mutacije su u Ashkenaza Jevreja, Kineza, Japanaca i Meksikanaca
Klinička slika. Kod osoba sa ovim tipom glikogenoze poremećena je produkcija glikoze putem glikogenolize, ali svakako i putem glikoneogeneze, i hipoglikemija se javlja na 3-4 sata poslije obroka. Laktična kisjelina, mokraćna kisjelina i trigliceridi su karakteristično povišeni, dok postoji neprikladna hipo- ketonemija u odgovoru na hipoglikemiju).
Simptomatska hipoglikemija i hepatomegalija su prvi znaci, koji se razvijaju u prvim mjesecima po rođenju, ali mogu da ostanu mjesecima skriveni. Hipoglikemije se ublažavaju čestim obrocima i na bolest se posumnja tek kada se poslije fizikalnog pregleda otkrije izrazita hepatomegalija. Simptomatska hipoglikemija se češće ispoljava kada se sa uzrastom djeteta povećava razmak izmedju obroka. Većina kliničkih simptoma ove bolest u vezi sa dva metabolička poremećaja: hipoglikemijom i laktičnom acidozom. Hipoglikemija je teška zato što su oštećeni i glikogenoliza i glikoneogeneza. Poremećena je i ketogeneza uprkos visokoj koncentraciji slobodnih masnih kisjelina. Hipoglikemija se posebno javlja kada je nivo insulina u krvi visok, kao u slučajevima naglog prekida intravenskog unosa glikoze, ili u slučaju kontinuiranog intragastričnog hranjenja. Nasuprot, pri hranjenju sa kukuruznim skrobom koji ima nizak glikemijski indeks, nivo insulina u krvi nije značajnije povišen. Hronično prisutna laktična acidoza prouzrokuje tahipneu, grčeve u mišićima i smanjenu podnošljivost fizičkih napora; može da se dešava i povraćanje usljed poremećaja gastričnog motiliteta.
Laboratorijski poremećaji koji se nalaze kod neliječene ili slabo liječene glikogenoze Tip Ia su: – metabolička acidoza koju prati povišeni anjonski gap, – hiperurikemija, – visok nivo transaminaza jetre (AST, ALT), – hiperlipidemija(posebno hipertrigliceridemija), – hiperlaktatemija (laktična acidoza), – hiperkalcemija, – povišeni inflamatorni markeri (sedimentacija i C-reaktivni protein), anemija, – trombocitoza, – produženo vrijeme krvarenja.
Dijagnoza. Najbrži dijagnstički postupak kojim se mogu dokazati poremećaji karakteristični za glikogenozu Tip Ia je brzi fasting test (3-4 sata). Kod Tipa Ia bolesti pri ovom testu će se javiti hipoglikemija i laktična acidoza. Pošto pri ovom testu glikemija može naglo da padne, treba biti oprezan, spreman za brzo reagovanje i izvoditi ga na opremljenom odjeljenju. Glikemiju treba mjeriti svakih 20-30 minuta poslije 2. sata. Kada glikemija padne na 50 mg/dl treba uzeti uzorak za odredjivanje laktata, a potom intravenski dati glukagon u dozi 0,03 ng/kg. Glukagon izaziva glikemijski odgovor i pogoršava laktičnu acidozu. Kada se test završi, treba dati intravenski glikozu da bi se uspostavila i održavala normalna koncentracija glikoze. Analizom mutacije gena definitivno se postavlja dijagnoza glikogenoze Tip Ia.









Add comment