
 Noonan sindrom je kongenitalno autozomno-dominantno oboljenje sa varijabilnom ekspresijom. Nastaje kao posljedica mutacije gena: PTPN11 (50% slučajeva), SOS1 (20%), RAF1 (10-15%), KRAS (5%). Tu može biti riječi o jednoj neotkrivenoj deleciji homolognog nosača X i Y hromozoma. Javlja se 1:1000 do 1:5000 novorođene djece. Poznat je pod različitim imenima: XY Turnerov fenotip, Familijarni Turnerov fenotip, Ullrichov sindrom. Dobio je naziv po američkoj doktorki (pedijatar-kardiolog) Noonan koja ga je prvi put opisala 1963. godine. Poremećaj je kombinacija hipogonadizma i karakterističnih somatskih anomalija, kao što su: anomalije lica, deformacija grudnog koša, urođene srčane mane, problemi sa vidom i sluhom, deformacije gastrointestinalnog sistema, genitouretalnog i koštano-mišićnog sistema.
Noonan sindrom je kongenitalno autozomno-dominantno oboljenje sa varijabilnom ekspresijom. Nastaje kao posljedica mutacije gena: PTPN11 (50% slučajeva), SOS1 (20%), RAF1 (10-15%), KRAS (5%). Tu može biti riječi o jednoj neotkrivenoj deleciji homolognog nosača X i Y hromozoma. Javlja se 1:1000 do 1:5000 novorođene djece. Poznat je pod različitim imenima: XY Turnerov fenotip, Familijarni Turnerov fenotip, Ullrichov sindrom. Dobio je naziv po američkoj doktorki (pedijatar-kardiolog) Noonan koja ga je prvi put opisala 1963. godine. Poremećaj je kombinacija hipogonadizma i karakterističnih somatskih anomalija, kao što su: anomalije lica, deformacija grudnog koša, urođene srčane mane, problemi sa vidom i sluhom, deformacije gastrointestinalnog sistema, genitouretalnog i koštano-mišićnog sistema.
Dijete rođeno sa tom anomalijom već prvih dana po rođenju, ako ima klasičnu formu bolseti, ispoljava znake koji su karakteristični za to oboljenje. Međutim, jedan broj te djece nema tipičnu kliničku sliku. Među njima se izdvaja grupa onih koji imaju manji broj malformacija ili su one slabije uočljive, tako da ih je teško dijagnostifikovati do puberteta, kada počinju da se ispoljavaju znaci hipogonadizma.
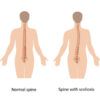
U adolescentno doba konstatuje se da je riječ o osobi malog rasta, specifičnog izraza lica sa: hipertelorizmom, antimongoloidnim rimama okuli, uskom gornjom čeljusti i mikrognatijom; vrat im je kratak i često sa prisutnom kožnom duplikaturom na spoljnjem dijelu vrata, koja polazi od mastoidnog vrha i širi se u vidu krilaca prema spoljnjem okrajku klavikule, tzv. pterygium colli. Takav vrat podsjeća na vrat sfinge a uši su široke i niske osnove pripoja. Povremeno se nalaze kifoza i skolioza, cubitus valgus gornjih i donjih ektremiteta. Čest je kritorhizam. Maljavost je oskudna i ženskog tipa, a nema je na grudima i licu. Kod njih često postoji anomalija krvih sudova, poremećaj metabolizma (šećerna bolest, giht) i druge promjene unutrašnjh organa.
Postavljanje dijagnoze
Detaljan objektivni pregled može da pobudi sumnju na ovaj sindrom. Međutim, definitivna dijagnoza se postavlja genetičkim testiranjem.
Paraklinički pokazatelji
Poslije puberteta konstatuje se hipergonadotropinemija (oba gonadotropina). Češće se konstatuje veća koncentracija FSH nego LH, jer su veće lezije u nivou spermatogeneze kao i niske koncentracije testosterona u plazmi. Rendgenografija kostiju i epifiznih hrskavica: znaci osteoporoze, nema srastanja epifiznih hrstavica, što je u disproporciji sa malim rastom.
Liječenje
Do puberteta, ukoliko je postavljena dijagnoza, primjenjuje se simptomatska terapija, kao i hirurško korektivno liječenje. Stimulacija rastenja, pri čemu se koristi humani hormom rastenja (genotropin, norditropin). Poslije puberteta, kada je ispoljen i kvantitativan i kvalitativan deficit polnih hormona, treba primijeniti androgene (testosteron) kojim se osoba maskulinizira do zadovoljavajućeg stepena.
Prikaz slučaja
Pacijent od 27 godina, javlja se na pregled izabranom ljekaru zbog: jakih bolova u lijevoj polovini vrata, otežanog gutanja, povraćanja, gubitka u težini (preko 10kg), oslabljenog sluha. Na osnovu anamnestičkih podataka i medicinske dokumentacije utvrđeno je da je u 4. godini rađena orhidopeksija lijevog testisa, u 14. godini orhidektomija zbog “atrofije lijevog testisa”. U 15. godini operisan zbog ASD-a srednjeg i proksimalnog septuma sa L-D šantom i stenozom A.Pulmunalis. Tada je utvrđena i hidronefroza lijevog bubrega. Rođen kao drugo dijete iz uredne trudnoće. Nije mu poznata visina majke i oca. Ne daje podatak o ranijoj mumps infekciji. Na pregledu pacijent tjelesne mase 49 kg, tjelesne visine 167 cm BMI 17,5, spušteni kapci, ušne školjke nisko usađene, gotsko nepce, kariozni zubi, Webbed neck, dekstrokonveksna skolioza, ograničena fleksija u struku, hiperelastična koža, hiperekstenzija zglobova. Grudni koš: pectus excavatum, lijevi skrotum prazan. U krvnoj slici normocitna anemija u hroničnoj bolesti (gladovanje), negativni zapaljenski sy. hipokreatinemija sa hiperurikemijom, hipoholesterolemijom, lako povećanim bilirubinom i ketonurijom (gladovanje), inicijalno subklinička hipertireoza (TSH manji od 0,01) sa kasnijom normalizacijom tiroidnih hormona, gonadotropini visoki (FSH 40,4; LH 12,6).
Nakon detaljnog hospitalnog ispitivanja na Klinici za Endokrinologiju i bolesti metabolizma KCS utvrđena je hemiagenezija desnog režnja štitaste žlijezde. Vrlo ekstenzivnim ispitivanjem: vrata, jednjaka i farinksa nije nađena organska lezija koja bi objasnila glavnu pacijentovu tegobu – bol u lijevoj polovini vrata zbog koje je u potpunosti prestao da unosi hranu.
Cilj rada
Skrenuti pažnju na nasljedno oboljenje koje često ostaje neprepoznato.
Zaključak
Pravovremenim postavljanjem dijagnoze na osnovu: anamnestičkih podataka – porodične anamneze, dijagnostičkih procedura i genetičkim ispitivanjem bi u mnogome olakšao kvalitet života osobe sa ovim sindromom.
Dr Bojana Vuković
Dom zdravlja, Bar





Add comment